Kardos Julianna
a kémiai tudomány doktora
A központi idegrendszer "validált" célfehérjéi
„tán jobb, hatékonyabb
fegyverek jobban szolgálnak, ha újra
összecsapunk,”
Milton, Elveszett paradicsom VI. [2]
Köszönöm a Magyar Tudományos Akadémia Biológiai, Kémiai és Orvosi Tudományok Osztályai tudós tagjainak érdeklődését és a megtisztelő lehetőséget néhány, az 1/047 sz. NKFP „Validált célmolekulákon alapuló gyógyszer- és diagnosztikum-tervezés (MediChem)” projekt (2001-2003) területén elért eredményünk ismertetésére.
Racionális gyógyszertervezés
A termék-ciklus idő optimalizálásának és rövidítésének igénye várhatóan jelentős változásokat idéz elő a gyógyszerfejlesztésben. Ennek a fejlesztési elvnek egyik lehetséges stratégiája a nagy áteresztőképességű módszerek (HTS) széleskörű alkalmazása. Ugyanakkor, a HTS módszerek alkalmazásával újabban nyert tapasztalatok fényében kezd általánossá válni az a nézet is, hogy a HTS módszerek kizárólagos alkalmazása veszélyeztetheti a gyógyszerfejlesztő szervezetek flexibilitását és versenyképességét, amennyiben gyengíti azon képességüket, hogy originális és innovatív gyógyszereket hozzanak létre. A HTS módszerekkel versenyezve, újabb lendületet vett a racionális gyógyszertervezés, amelynek technikai bázisa a molekuláris biológiai módszerek, a szintetikus és analitikai eszköztárak valamint az adatfeldolgozás és tárolás folyamatos fejlődése és alkalmazása.
Összehasonlító molekulatér analízisen alapuló kvatitatív szerkezet-hatás modellek (QSAR/CoMFA) alkalmazása a racionális gyógyszertervezésben
A racionális gyógyszertervezés egyik lehetséges megközelítési módja,
a gyógyszerjelölt molekulák szerkezet-hatás összefüggéseinek felismerésén
alapuló QSAR/CoMFA farmakofór modell. A további szintetikus erőfeszítéseket
megalapozó QSAR/CoMFA farmakofór modellek sikeresen alkalmazhatóak gyógyszerjelöltek
receptor és enzim altípusokkal szemben mutatkozó specificitásának előrejelzésére
([3], 1. Táblázat).
| Célmolekula: Receptor | Célmolekula: Enzim |
| Dopamin D1 vs D2 | MAO-A vs MAO-B |
| GABA vs Benzdiazepin | MMP-8 vs MMP-3 |
| 5HT1A vs a1 | Tripszin vs Trombin vs Faktor Xa |
| PNMT vs a2 | |
| 5HT2A vs Dopamin D2 | |
| 5HT2B vs 5HT2C | |
| 5HT vs NA vs DA | |
| Ösztrogén a vs b | |
| Ópioid d vs m | |
| 1. Táblázat. A QSAR/CoMFA módszer néhány sikeres alkalmazása célfehérje altípusok közötti szelektivitás meghatározására ([3]: Podlogar, Brent L. és Ferguson, David M.: QSAR and CoMFA: A perspective on the practical application to drug discovery. Drug Design and Discovery. 17 (2000) 1. 4-12. p. nyomán) | |
A célmolekula szerkezetének ismeretén alapuló racionális gyógyszertervezés: a 3D farmakofór modell
A másik lehetséges megközelítési mód, a validált célmolekulákon alapuló
racionális gyógyszertervezés, amely a célmolekula 3D szerkezetének ismeretén
illetve új célmolekulák felismerésén és 3D szerkezetének meghatározásán
alapul. Néhány - az idegi jelátvitelben kulcsfontosságú – receptor, ioncsatorna
és enzim szabad valamint specifikus ligandumával kölcsönható szerkezete
vált ismertté a közelmúltban (Brookhaven Data Bank, 2. és 3. Táblázat).
Így lehetőség nyílik arra, hogy a biológiai hatásáért felelos - a gyógyszerjelölt-célmolekula
kölcsönhatás eredményeképpen kialakult - globális és lokális szerkezeti
változásokat, a kisérleti 3D farmakofórt megismerhessük.
|
|
||||
| nACh receptor | Patkány, M2 hélix |
|
|
1CEK |
| NMDA receptor | Humán, M2 hélix (1NR1) |
|
|
1CEK |
| Patkány iGluR2 S1S2*Glu |
|
|
|
|
| Patkány iGluR2 S1S2*kainát |
|
|
|
|
| Patkány iGluR2 S1S2*DNQX |
|
|
|
|
| Patkány iGluR2 S1S2*AMPA |
|
|
|
|
| Patkány iGluR2 S1S2 apo |
|
|
|
|
| Patkány iGluR2 S1S2*kainát |
|
|
|
|
| K+ csatorna | Patkány, intracelluláris domén |
|
|
|
|
|
||||
| mGluR1 | Patkány, extracelluláris domén*Glu |
|
|
|
| Patkány, extracelluláris domén,
szabad forma I |
|
|
|
|
| Patkány, extracelluláris domén,
szabad forma II |
|
|
|
|
| Fotoreceptor | Marharetina, rodopszin
+11-cis retinal |
|
|
1HZX |
| 2. Táblázat. Plazmamembrán receptorok és ioncsatornák 3D Szerkezetére vonatkozó adatok. Rövidítések: NMR - magmágneses rezonancia spektroszkópia; XD - röntgenkrisztallográfia; a célfehérje méretét az aminosav oldalláncok száma mutatja; a megadott szám és betűkód alapján azonosítható a kisérleti szerkezet a Brookhaven Data Bankban tárolt adatfájlok között; 7TM receptorok - hét transzmembrán hélixből álló membránproteinek, melyek intracelluláris doménje specifikus G protein heterotrimérhez kapcsolódik. | ||||
|
|
||||
| ATP szintáz | Szív, F1F0 ATPase |
|
3248 |
1COW, 1BMF |
| GTP hidroláz
(G protein heterotrimér) |
Agy, Gia1GDP
Agy, Gia1GTP Kiméra, Gta/iaGDP*Gtßg Retina, GtaGDP+Mg2+ Retina, GtaGTP |
XD XD XD XD XD XD XD XD XD |
605 353 353 764 763 650 972 324 1632 |
1BKD 1GDD 1CIP 1GG2, 1GP2, 1GOT 1A0R 1TAD 1TND 1TAG 1TBG |
| 3. Tálázat. Néhány, az idegi jelátvitelben kulcsfontosságú enzim 3D szerkezetére vonatkozó adatok. (NMR: magmágneses rezonancia spektroszkópia, XD: röntgen-krisztallográfia, a célfehérje méretét az aminosav oldalláncok száma mutatja, megadott szám és betűkód alapján azonosítható a kisérleti szerkezet a Brookhaven Data Bankban tárolt adatfájlok között.) | ||||
Néhány - az idegi jelátvitelben kulcsfontosságú - célmolekula szabad (apo) valamint specifikus ligandumával kölcsönható szerkezete.
Receptor ioncsatornák
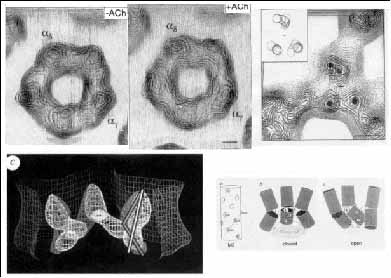
1. ábra. A zárt és nyitott állapotú ioncsatornát formáló nikotinos acetilkolin receptor (nAChR) heterooligomer konformációi. A pentamer nAChR extracelluláris doménjéhez kötődő agonista (ACh) hatására bekövetkező konformációváltozás (v.ö. elektronsűrűség eloszlási térképek) laterális konformációváltozásokat indukál, amelyek vertikálisan terjednek tovább. Ennek következménye, hogy az ioncsatorna falát képező transzmembrán hélixek (M2) zárt állapotát konstituáló Leu-gyűrű nagyobb átmérőjű körpályára kerül, az ioncsatorna átmenetileg kinyílik (alsó képsor). Feltételezhető, hogy az nAChR alegységekkel homológ szerkezetű alegységekből felépülő gamma-aminovajsav GABAA, glicin és 5-OH-triptamin (5-HT3) ioncsatornát formáló receptorok hasonló mechanizmussal aktiválódnak ([4]: Unwin, N. Acetylcholine receptor imaged in the open state. Nature. 373 (1995) 37-43 p. nyomán)
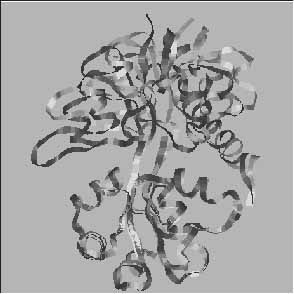
2. ábra. Az ioncsatornát formáló Glu receptorok családjába tartozó AMPA típusú receptor heterooligomér iGluR2 alegység extracelluláris kötődomén globális konformációváltozása agonista (Glu) jelenlétében (fekete) és távollétében (szürke). Megállapítható, hogy Glu jelenlétében az extracelluláris kötődomén zártabb alakzatot vesz fel. A feltételezés szerint, ennek következtében („Venus Flytrap” mechanizmus) távolabb kerülnek egymástól az ioncsatorna falát képező transzmembrán hélixek és az ioncsatorna kinyílik ([5]: Armstrong, N. és Gouaux, E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structure of the GluR2 ligand binding core. Neuron. 28 (2000) 10. 165-181. nyomán)
 |
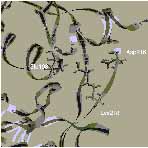 |
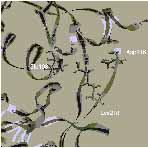
3. ábra. Az ioncsatornát formáló Glu receptorok családjába tartozó AMPA típusú receptor heterooligomér iGluR2 alegység extracelluláris kötődomén szerkezeti részlete (szürke): egy lokális változás, a lizin-kapcsoló állása antagonista (bal, fekete) és agonista (jobb, fekete) jelenlétében. A molekulamechanikai számítások szerint, a lizin218 legalacsonyabb energiájú állapota az agonista és antagonista karakterű ligandumokkal együtt kristályosított szerkezetben talált orientációnak felel meg. Az agonista és antagonista karakterű ligandumokat az "üres" (apo) szerkezetbe dokkolva a lizin218 kapcsoló állása molekulamechanikai számításokkal reprodukálható. Az apo-szerkezeten alapuló homológia modell olyan agonista és antagonista iGluR2 kötődomén komplexek esetében is alkalmazható, amelyekre vonatkozóan nem áll még rendelkezésre a kristályos szerkezet. ([6]: Nyikos Lajos, Simon Ágnes, Barabás Péter, Kardos Julianna. Ligand-specific conformations of an ionotropic glutamate receptor. Protein Engineering, 2002, közlésre beküldött kézirat nyomán)A globális konformációváltozásokat tükröző, lokális szerkezeti változások felismerésével új - a célmolekulákhoz kötődő - vegyületek várható biológiai hatása molekulamechanikai számításokkal is tervezhető [6-8]. Ebben a megközelítési módban a szintézisre vagy funkcionális vizsgálatokra kijelölhető molekulák köre gyorsan és jelentősen leszűkíthető.
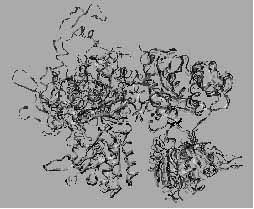
4. ábra. A G proteinnel kapcsolt, hét transzmembrán hélix szakaszból álló (7TM) membránreceptorok „ C” családjába tartozó metabotróp Glu receptor (mGluR1) dimér Glu távollétében felvett két különböző konformációja (szabad I: sötétszürke, szabad II: szürke) valamint a Glu jelenlétében meghatározott konformáció (fekete) összehasonlítása. Megállapítható, hogy a szabad II és az mGluR1*Glu komplex konformációk hasonlóak, ugyanakkor kölcsönösen eltérnek a szabad I konformációtól. Ennek alapján felvethető, hogy a kisérletileg tapasztalt szabad II állapot konstitutíve aktív. Ebben az esetben az agonista funkciója az, hogy az aktív szabad II konformációt stabilizálja. ([9]: Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 407 (2000) 971-977 p. nyomán)
7TM receptorok
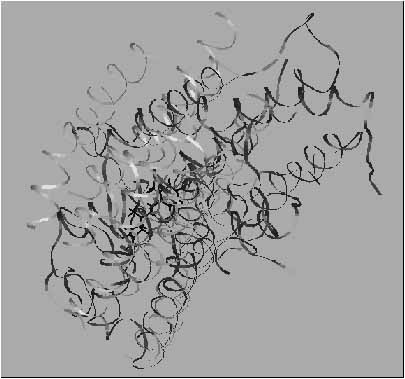
5. ábra. A fény hatására aktiválódó, 11-cisz-retinált tartalmazó 7TM receptor - a rodopszin - modellje (szürke) és kristályszerkezete (fekete). Megállapítható, hogy a hasonló felépítés ellenére, a 11-cisz-retinál kötőhelye nem teljesen azonos a modellben és a kristályban, a modell szerkezet és a kisérleti szerkezet jelentősen különbözik. Ennek egyik oka, hogy a kisérleti szerkezet a transzmembrán hélixeket összekötő szakaszokat is tartalmazza, míg a modell nem. Egy másik lehetséges ok, hogy mivel a felszíni aminosav oldalláncok a kristá-lyos kontaktusok képződése során rendeződnek, a kristályos szerkezet kialakulása önmagában konformációs entrópia-változásokakal jár együtt. Ehhez adódnak hozzá a ligandum kötődésé-nek hatására létrejövő globális konformációs entrópia-változások. Az entrópia-változásokat nem lehet számolni, éppen ez indokolja a célmolekulák és specifikus ligandumaikkal alkotott komplexeik kisérleti szerkezetén alapuló gyógyszertervezésre irányuló erőfeszítéseket. Ugyanakkor, a röntgenkrisztallográfiai szerkezetükkel validált célfehérjék esetében általánosan felmerülő kérdés az, hogy az izolált protein kristályosodása eredményeképpen létrejövő szerkezet mennyiben tekinthető autentikusnak, mennyiben felel meg a membránba ágyazott natív protein szerkezetének? ([10]: Pogozheva, I.D., Lomize, A.L., Mosberg, H.I. The transmembrane 7-alpha-bundle of rhodopsin: distance geometry calculations with hydrogen bonding constraints. Biophys. J. 72 (1997) 1963-1985 p. valamint [11]: Palczewski, K., Kumasaka. T., Hori, T., Behnke, C.A., Motoshima, H., Fox, B.A., Le Trong, I., Teller, D.C., Okada, T., Stenkamp, R.E., Yamamoto, M., Miyano, M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 289 (2000) 739-745 p. nyomán)
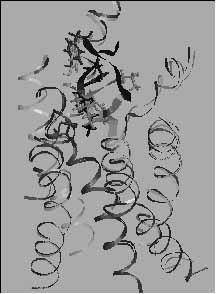
6. ábra. A kristályos rodopszin szerkezet alapján [11] felépített szomatosztatin receptor (SSTR2) homológia modell. A szomatosztatin receptor modell és a neuropeptid szomatosztatin (SST: Ala-Gly-Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys) (fekete) valamint egy - tumorgátló hatású [12] - szomatoszatin analóg (TT2-32: D-Phe-Cys-Tyr-D-Trp-Lys-Cys-Thr-NH2) kötődési kölcsönhatását (szürke) molekulamechanikai számításokkal vizsgáltuk. A számítások szerint a TT2-32 erősebben kötődik az SSTR2 modellhez, mint az SST, amely megmagyarázhatja e vegyületek receptor-kötődési és funkcionális mérésekben mutatkozó különbségét. A modellezéshez a Swiss-Model szervert alkalmaztuk [13]. A templát keresésekor a BLAST programmot alkalmaztuk. Az SSTR2 molekula feltételezett helikális szakaszait transz-membrán szakaszokat becslő HMMTOP módszerrel jelöltük ki [14].
 |
 |
7. ábra. Egy G protein heterotrimér, a Gta/iaGtbg kristályos szerkezete. Sejtfelszíni 7TM receptorok százai, mint például a szaglás, ízlelés, látás, jelátvitel (ACh, Glu, GABA, adrenalin, dopamin, szerotonin, peptidek és hormonok) receptorai „nanogépekként” működnek, amelynek nyomógombja a 7TM receptor, kapcsolója a Ga és propellere a Gbg. A specifikus ligandum (vagy fény) és 7TM receptor klcsönhatás eredményeképpen a 7TM receptor intracelluláris doménjének konformációja megváltozik és ez felgyorsítja a GDP disszociációját a 7TM receptorhoz kapcsolódó Ga alegységről A ledisszociált GDP helyére GTP kötődik. A GTP kötődés hatására bekövetkező konformációváltozás (v.ö. 8. ábra) idézi elő a heterotrimér disszociációját Ga és Gbg komponenseire. A disszociált komponensek különböző effektorokat (enzimek és ioncsatornák) aktiválnak. A Ga alegység valójában egy GTP hidroláz, amely a hozzá kötődő GTP-t GDP-re bontja. Az így képződő Ga*GDP komplex újból egyesül a Gbg komponenssel és a képződő heterotrimér a 7TM receptorával. Ezzel visszajutunk a kezdeti állapothoz, és a 7TM receptor ismét aktiválható. ([5]: Clapham, D. E. The G-protein nanomachine. Nature. 379 (1996) 397-399. p. és [16]: Lambright, D.G., Sondek, J., Bohm, A., Skiba, N.P., Hamm, H.E., Sigler, P.B. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 379 (1996) 311-319. p. nyomán)
 |
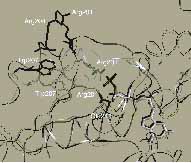 |
8. ábra. A rodopszin receptorhoz kapcsolt G protein heterotrimér Gta alegységének kristályos szerkezete GDP és Mg2+ (szürke) valamint egy nem hidrolizáló GTP analóg (fekete) jelenlétében. A balodalon a globális szerkezet, a jobboldalon pedig az a szerkezeti részlet látható, amelynek konformációváltozása előidézi a heterotrimér disszociációját Gta és Gtbg komponenseire[17]: Lambright, D.G., Noel, J.P., Hamm, H.E., Sigler, P.B. Structural determinants for activation of the alpha-subunit of a heterotrimeric G protein. Nature. 369 (1994) 621-628. p. és [18]: Noel, J.P., Hamm, H.E., Sigler, P.B. The 2.2 A crystal structure of transducin-alpha complexed with GTP gamma S. Nature. 366 (1993) 654-663. p. nyomán)
A módszerek felhasználása
Az új, várhatóan neuroprotektív hatású vegyületek tervezésére javasolt
módszereket az 1/047 NKFP „Validált célmolekulákon alapuló gyógyszer- és
diagnosztikum-tervezés” (MediChem) projekt 3. számú részfeladatának kidolgozásában
alkalmazzuk.
Köszönetnyilvánítás
A bemutatott eredményekért nemcsak Nyikos Lajosnak és Simon Ágnesnek tartozom köszönettel hanem az MTA Kémiai Kutatóközpont Neurokémiai Osztály valamennyi munkatársának. Külön köszönöm az 1/047 sz. NKFP "Validált célmolekulákon alapuló gyógyszer- és diagnosztikum-tervezés (MediChem)" projekt szakmai vezetőjének, Szántay Csaba akadémikusnak és a MediChem Konzorcium két témafelelősének, Dr. Juhász Gábornak és Kéri György professzornak közreműködését valamint a MediChem Konzorcium vezető intézményének, az MTA Kémiai Kutatóközpontnak az erkölcsi és anyagi támogatást.
Bibliográfiai leírás
1. The Compact Edition of Oxford Dictionary. (1971) Oxford.
2. Milton válogatott művei. Elveszett paradicsom. VI. Könyv. (1978) 211. p. Európa, Budapest.
3. Podlogar, Brent L. és Ferguson, David M.: QSAR and CoMFA: A perspective on the practical application to drug discovery. Drug Design and Discovery. 17 (2000) 1. 4-12. p.
4. Unwin, N. Acetylcholine receptor imaged in the open state. Nature. 373 (1995) 37-43. p.
5. Armstrong, Neali és Gouaux, Eric: Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structure of the GluR2 ligand binding core. Neuron. 28 (2000) 10. 165-181. p.
6. Nyikos Lajos, Simon Ágnes, Barabás Péter, Kardos Julianna. Ligand-specific conformations of an ionotropic glutamate receptor. Protein Engineering, 2002, közlésre beküldve.
7. Szárics Éva, Nyikos Lajos, Barabás Péter, Kovács Ilona, Skuban Nina, Temesváriné-Major Eszter, Egyed Orsolya, Nagy, Peter I., Kökösi József, Takács-Novák Krisztina és Kardos Julianna: Quinazolone-alkyl-carboxylic acid derivatives inhibit transmembrane Ca2+ ion flux to (+)[S]-a-amino-3-hydroxy-5-methylisoxazole-4-propionic acid. Molecular Pharmacology. 59 (2001) 4. 920-928. p.
8. Keserű György M. és Kolossváry I.: Molecular Mechanics and Conformational Analysis in Drug Design (Blackwell Science, Oxford, 1999).
9. Kunishima, N., Shimada, Y., Tsuji, Y., Sato, T., Yamamoto, M., Kumasaka, T., Nakanishi, S., Jingami, H., Morikawa, K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 407 (2000) 971-977. p.
10. Pogozheva, I.D., Lomize, A.L., Mosberg, H.I. The transmembrane 7-alpha-bundle of rhodopsin: distance geometry calculations with hydrogen bonding constraints. Biophys. J. 72 (1997) 1963-1985. p.
11. Palczewski, K., Kumasaka, T., Hori, T., Behnke, C.A., Motoshima, H., Fox, B.A., Le Trong, I., Teller, D.C., Okada, T., Stenkamp, R.E., Yamamoto, M., Miyano, M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 289 (2000) 739-745. p.
12. Kéri, Gy., Érchegyi, J., Horváth, A., Mező, I., Idei, M., Vántus, T., Balogh, Á., Vadász, Zs., Bökönyi, Gy., Teplán, I., Csuka, O., Tejeda, M., Gaál, D., Szegedi, Zs., Szende, B., Roze, C., Kalthoff, H., Ullrich, A. Tumor-selective somatostatin analog (TT-232) with strong in vitro and in vivo antitumor activity. Proc. Natl. Acad. Sci. (USA). 93 (1996) 12513-12518. p.
13. Guex, N., Peitsch, M.C., SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modelling. Electrophoresis.18 (1997) 2714-2723. p.
14. Tusnády, G.E, Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 17 (2001) 9. 849-850. p.
15. Clapham, D. E. The G-protein nanomachine. Nature. 379 (1996) 397-399. p.
16. Lambright, D.G., Sondek, J., Bohm, A., Skiba, N.P., Hamm, H.E., Sigler, P.B. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 379 (1996) 311-319. p.
17. Lambright, D.G., Noel, J.P., Hamm, H.E., Sigler, P.B. Structural determinants for activation of the alpha-subunit of a heterotrimeric G protein. Nature. 369 (1994) 621-628. p.
18. Noel, J.P., Hamm, H.E., Sigler, P.B. The 2.2 A crystal structure
of transducin-alpha complexed with GTP gamma S. Nature. 366 (1993) 654-663.
p.
Rezümé
The need to optimise and shorten the time for the product-cycle may alter significantly the drug development. In meeting this principle, one of the possible strategy has relied upon a wide application of high throughput methodologies (HTS). Nevertheless, experiences recently obtained with the use of HTS made now becoming accepted the view that the sole reliance on HTS may actually threat the pharmaceutical organization's flexibility and competitiveness as HTS weaken the organization's ability to create original and innovative therapeutics.
These realities raised the opportunities - in an ongoing competition with HTS - for a resurgence of rational drug design. These are based technically on the continuous progress and application of molecular biology, new synthetic and analytical tools and data analysis and management. One of the approaches of rational drug design is the extension of the quantitative structure-activity relationships to the method of comparative molecular field analysis (QSAR/CoMFA), i.e. the use of the bioactive conformation of the ligand. The most active use of the QSAR/CoMFA pharmacophore model is to address issues of target (receptor and enzymes subtypes) selectivity that would guide synthetic efforts.
The other approach of rational drug design is based on validated targets,
i.e.
the knowledge of the experimental structure of ligand-target co-complexes
including the discovery and structural validation of new targets. The 3D
structure of several receptors and enzymes of key importance in neural
signalling has been known recently (Brookhaven Data Bank). Thus provides
the opportunity to disclose global and local conformational changes sequential
to ligand-target interactions that are responsible for the biological effect.
The knowledge of local structural changes reflecting global conformational
transitions enables us to predict the potential biological effect of new
ligands by computational approaches such as ligand-docking. The approach
adds to the drug discovery toolbox the value of fast and significant reduction
in the number of new molecules to synthesise and/or to test.
| Gyógyszerkutatás: jelen és jövő | http://www.kfki.hu/chemonet/
http://www.chemonet.hu/ |