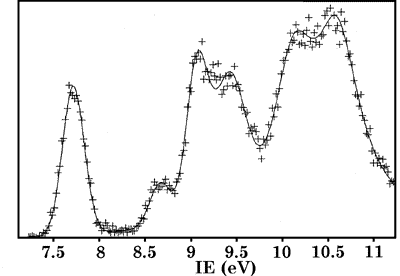
Figure 1. The photoelectron spectrum of Me4Sb2
Sztáray Bálint
Fémorganikus vegyületek ionizált állapotai és bomlása
Eötvös Loránd Tudományegyetem
Budapest
2000
Bevezetés
A diffrakciós eljárások mellett a molekulaspektroszkópiai módszerek segítségével tanulmányozhatjuk leghatékonyabban kísérleti úton a molekulák elektron- és térszerkezetét. A vákuum ultraibolya fotoelektron-spektroszkópia (Photoelectron Spectroscopy, PES; Ultraviolet Photoelectron Spectroscopy, UPS) segítségével a vizsgált rendszer vegyértékelektronjaihoz tartozó ionizációs energiákat mérhetjük, melyeket a Koopmans-elv segítségével a molekulapályák energiáival hozhatunk kapcsolatba. A fémorganikus vegyületek elektronszerkezete különös érdeklõdésre tarthat számot a spektroszkópusok között, ezeknél ugyanis a szerves vegyületekkel ellentétben általában jól elkülöníthetõek a különbözõ kötéstípusokhoz rendelhetõ molekulapálya-energiák. A klasszikus He(I) és He(II) fotoelektron-spektroszkópia ugyan már nem tekinthetõ igazán modern vizsgálati módszernek, azonban jelenleg is még a leghatékonyabb, és -pontosabb eszköz különféle konformációs problémák megoldására.
A fotoelektron-spektroszkópia testvérmódszerének tekinthetõ a küszöb fotoelektron-fotoion koincidencia- (Threshold Photoelectron Photoion Coincidence, TPEPICO) spektroszkópia, mely a fotoionizáció során keletkezõ elektronok kinetikusenergia-analízisén túlmenõen az egyidejûleg keletkezõ ionok tömegeloszlását is vizsgálja. Gyakorlatilag a módszer a tömegspektrometria rendkívüli pontosságú változatának tekinthetõ, segítségével belsõ energia szerint kiválasztott ionok unimolekulás bomlása vizsgálható. Mivel a fémorganikus molekulák termokémiája az utóbbi években rendkívüli fontosságra tett szert (köszönhetõen elsõsorban a fémorganikus katalizátorok elterjedésének), a fémorganikus TPEPICO az elkövetkezõ években jelentõs karriert futhat be. Különösen érdekes a fémorganikus molekulákból bizonyos, a katalítikus elemi lépések szempontjából is fontos ligandumok bomlásainak kinetikája, melynek mérését e spektroszkópia szintén lehetõvé teszi.
Jelen doktori értekezés három témakörre osztható fel. Elsõként az AsAs és SbSb kötést tartalmazó vegyületeket tárgyalom, majd egy tetrahidridoborát-komplex következik. Az utolsó témakör pedig fémorganikus vegyületek TPEPICO-vizsgálatát foglalja magában. Némileg kakukktojásként itt egy szerves molekuláról is ejtek néhány szót, ez ugyanis a spektroszkópiai eredmények modellezése szempontjából volt számomra különösen érdekes.
Arzén- és antimonorganikus vegyületek
A hidrazinban és szubsztituált származékaiban a szomszédos N-atomok magános párjai döntõ mértékben befolyásolják a molekulák sztereokémiai tulajdonságait. Ha a két magános pár között jelentõs a kölcsönhatás, akkor a pályák szimmetrikus (n+) és antiszimmetrikus (n) kombinációjának az energiája különbözõ. Jelen munkában a tetrametil-hidrazin arzén- és antimonanalógjának elektronszerkezetét és rotációs izomériáját tanulmányoztuk.
Cowley és Dewar vizsgálatai alapján a Me4P2 és a Me4As2 fotoelektron-spektrumában észlelhetõk mind az anti-, mint pedig a gauche-konformerhez rendelhetõ sávok. A Me4Sb2 általunk felvett és publikált spektruma a foszfor- és az arzénanalógok spektrumához nagyon hasonlít. Mivel a spektrumnak a fenti irodalommal analóg értelmezése ellentmondott az általunk elvégzett kvantumkémiai számítási eredményeknek, részletesen vizsgáltuk különféle ab initio szinteken a Me4As2 és a Me4Sb2 tér- és elektronszerkezetét. Ezek a számítások segítettek megoldani a fotoelektron-spektrumok értelmezésénél felmerült problémákat, valamint érdekes kvalitatív eredményekre is vezettek a magános párok energetikájával és a térszerkezetre gyakorolt hatásával kapcsolatban.
Bisz-tetrahidridoborát-bisz-ciklopentadienil-cirkónium(IV)
Az átmenetifém-tetrahidridoborátok tér- és elektronszerkezete az utóbbi években nagy érdeklõdést váltott ki. Ezek a vegyületek praktikus szempontból is érdekesek, mind katalizátorként, mind pedig gõzfázisú kémiai leválasztás prekurzoraként való alkalmazás szempontjából. Elméletileg elsõsorban a fémcentrum és a tetrahidridoborát-ligandum kapcsolódása érdekes, amely megvalósulhat egy, két, illetve három hidrogénhídon keresztül is. Elektron-, valamint röntgendiffrakciós módszerekkel a hidrogénatomok helyzete csak nagy bizonytalansággal határozható meg. Azonban a diffrakciós módszerekkel mért fém-bór távolságokból a kapcsolódási mód megbecsülhetõ, hiszen más effektív sugár rendelhetõ a különbözõ kapcsolódású tetrahidridoborát-ligandumokhoz. Újabban pedig kvantumkémiai számítások segítségével lehetséges direkt elméleti információt nyerni a molekulageometriáról; ilyen esetben más számított paramétereknek a kísérletileg mérhetõkkel való összevetése fontos lehet a számítások ellenõrzéséhez. A vegyület elektronszerkezete, molekulapálya- és ionizációs energiák tekintetében például a fotoelektron-spektroszkópia és a kvantumkémia egymást segítõ, kiegészítõ és ellenõrzõ módszernek alkalmas.
Fotoelektron-fotoion koincidenciaspektroszkópia
Átmenetifémek és átmenetifém-komplexek számos ipari jelentõségû reakcióban használatosak katalizátorként. Ezek hatékonysága sokféle tényezõ függvénye, melyek közül a központi fém hozzáférhetõsége igen lényeges. A központi fém leggyakrabban egy viszonylag lazán kötött ligandum disszociációjával válik támadhatóvá, tehát e folyamat energetikájának, valamint kinetikájának ismerete igen fontos a jövõ katalizátorainak megtervezéséhez. Az általunk vizsgált ciklopentadienil-kobalt-dikarbonil, valamint a ciklopentadienil-mangán-trikarbonil például sok katalizátor alapvegyületei, így termokémiájuk vizsgálata komoly érdeklõdést keltett már eddig is. Sajnos azonban eddig csak kis pontosságú mérési eredmények hozzáférhetõek a szakirodalomban; ez a helyzet egyébként a legtöbb hasonló fémorganikus komplex esetében. Az általunk elvégzett TPEPICO-mérésekben a karbonilvesztések energetikáját, és kinetikáját vizsgáltuk. Bebizonyosodott, hogy az utóbbi az elõbbinek elõfeltétele, hiszen pontos energetikai adatok csak a sebességi állandók megfelelõ modellezésével nyerhetõk. Ugyancsak fontos konszekutív bomlások során a termékek energiaeloszlásának modellezése.
A kompetitív disszociáció modellezése is igen érdekes és fontos. Ennek illusztrálására közlöm a jódacetonitril TPEPICO-spektrumát, és ennek kiértékelését. Ez a molekula a legnagyobb jóindulattal sem nevezhetõ fémorganikusnak, tehát az értekezésbõl kilóg. Azért tartom mégis fontosnak, hogy jelen disszertációban közöljem a vele kapcsolatos eredményeinket, mert TPEPICO-spektrumai két kompetitív bomlást mutatnak, így ezek pontos kiértékelése igen érdekes feladat. Amennyiben ugyanis két disszociációs folyamat aktiválási energiája közel esik egymáshoz, a második leányion megjelenési energiája a letörési görbébõl közvetlenül csak igen pontatlanul kapható meg. A fragmensek képzõdéshõinek meghatározásához azonban értelemszerûen ez az adat nagyon lényeges. A CH2CN-gyök és -ionok termokémiájának felderítése pedig gyakorlati szempontból is igen fontos. Számos magas hõmérsékletû folyamatban lehetnek ugyanis átmeneti termékek az itt tárgyalt gyökök és ionok. Ezeknek a folyamatoknak a különféle égési folyamatok leírásánál fontos modellezéséhez magától értetõdõ módon szükség van pontos termokémiai adatokra. Nem véletlen tehát, hogy például gázfázisú ionkémiai konferenciákon lépten-nyomon a Légierõ kutatóiba botlik a résztvevõ.
Eredmények
Magánospár-kölcsönhatás
Bisz-tetrahidridoborát-bisz-ciklopentadienil-cirkónium(IV)Felvettük a tetrametil-distibán fotoelektron-spektrumát. Kvantumkémiai számításokat végeztünk a Me4As2 és a Me4Sb2 molekulákra. Elméletileg bizonyítottuk a gauche- és az anti-rotamerek létezését. Meghatároztuk a két molekula konformereinek ab initio geometriáját HF, MBPT(2) és CCSD szinten. Kiszámítottuk e két molekula potenciális energiájának torziótól való függését. Kiszámítottuk az arzén és az antimon magános párok HF-pályaenergiáit a különbözõ rotamerekben. Meghatároztuk a magánospár-felhasadás torziófüggését. EOMIP-CCSD számításokat végeztünk az ionizációs energiák nagyon pontos meghatározására. Fentiek alapján újraértelmeztük a Me4As2 Cowley és munkatársai által publikált fotoelektron-spektrumát. A spektrumok új asszignációjának megfelelõen meghatároztuk a konformerarányokat, ezeket összevetettük a számított és az eddig publikált adatokkal. Megállapítottuk, hogy az elméleti és a fotoelektron-spektrumból számítható konformerarány közel áll egymáshoz, ettõl az elektrondiffrakcióból kapott érték jelentõsen eltér. A magánospár-felhasadás Dewar-féle értelmezése helyett új magyarázatot adtunk a jelenségre, mely figyelembe veszi e molekulapályák s-pálya karakterét is.
Felvettük a molekula fotoelektron-spektrumát He(I) és He(II) fotonenergiánál. Négy, egymástól jól elkülönülõ sávrendszert tudunk megkülönböztetni. Az elsõ 8.5-9.6 eV nagyságú ionizációs energiához tartozik, a második 10-11.5 eV-nál, a harmadik, legintenzívebb sáv 13-14 eV-nál, és végül a negyedik 17 eV környékén található.CpCo(CO)2A vegyületre teljes geometriaoptimalizálást végeztünk a Turbomole programcsomag használatával zárt héjú HartreeFock szinten Pulay-féle természetes koordinátákban. A vegyület számított térszerkezete a két hidrogénhidas szerkezetet igazolta. A Cp-gyûrûk egymáshoz képest elfordult helyzetben állnak, így a vegyületnek nincs teljes C2v szimmetriája.
A számított elektronszerkezetre populációanalízist végeztünk. E szerint a legfelsõ pályarendszerben, valamint a középsõ sáv legmagasabb energiához tartozó molekulapályájában jelentõs a d-pálya részvétel. A gyakorlati tapasztalatok alapján ezekhez a pályákhoz tartozó ionizációnál intenzitásnövekedésnek kell jelentkeznie a He(II)-spektrumban. Ez a mért spektrummal nagyon jó egyezésben van, az elsõ sáv nagymértékû intenzitásnövekedése, valamint a középsõ sáv alakjának megváltozása igazolja a d-pálya részvételt.
Rokon vegyületek spektruma, valamint a számítások alapján megállapítottuk, hogy az elsõ sáv a ciklopentadienil-cirkónium kötéshez tartozik, a második sáv a tetrahidridoborát-ligandumhoz és tetrahidridoborát-fém kötéshez tartozó ionizációhoz kapcsolható, a harmadik a ciklopentadienil-ligandumról, míg a negyedik a ciklopentadienil-cirkónium kötõpályákról történõ ionizációhoz rendelhetõ.
A vegyértékhéj-ionizációs energiák pontos meghatározására a körülbelül harmadrendig pontos OVGF-módszerrel végeztünk számítást, a kapott ionizációs energiák a kísérletivel igen jól egyeznek.
Röntgendiffrakciós vizsgálatokkal meghatároztuk a vegyület szilárd fázisú szerkezetét, a tetrahidridoborát-ligandum hidrogénjeinek a pozíciója ugyan nem állapítható meg pontosan, de a cirkónium-bór távolság a számításokkal egybehangzó módon kétfogú ligandumra utal.
Felvettük a ciklopentadienil-kobalt-dikarbonil (CpCo(CO)2) küszöb fotoelektron-spektrumát. A meghatározott elsõ ionizációs energia az irodalmi, a He(I) és He(II) spektrum alapján közölt adattal jó egyezésben van.CpMn(CO)3A vegyületre TPEPICO-vizsgálatokat végeztünk, meghatároztuk a CpCo(CO)2+-ion karbonilvesztéseihez tartozó letörési görbéket, valamint különbözõ fotonenergiáknál a repülésiidõ (TOF)-tömegspektrumot.
Különbözõ hõmérsékleteken megmérve a minta gõznyomását, meghatároztuk a vegyület párolgáshõjét.
Kvantumkémiai számításokat végeztünk a vegyületre, valamint a belõle képzõdõ ionokra HartreeFock és MBPT(2) szinten, valamint sûrûségfunkcionál-elmélettel B3LYP-funkcionált használva. Meghatároztuk az egyensúlyi szerkezeteket és a hozzátartozó rezgési frekvenciákat.
A CpCo(CO)2+-ion karbonilvesztéséhez tartozó reakciókoordináta mentén kiszámítottuk a potenciálisenergia-görbét, ennek mentén pedig a rezgési frekvenciák változását.
A variációs átmenetiállapot-elmélet (VTST) segítségével meghatároztuk az átmeneti állapot közelítõ helyét.
A lejátszódó konszekutív unimolekulás karbonilvesztési folyamatokat az RRKM-elmélet segítségével, a számított rezgési frekvenciák felhasználásával modelleztük. Mind a TOF-, mind a letörési görbéket a kísérleti adatokhoz illesztve meghatároztuk a disszociációkhoz tartozó gátmagasságokat, valamint az átmeneti állapotok közelítõ aktiválási entrópiáját.
Meghatároztuk a disszociációs folyamatok sebességi állandóját, valamint ezek energiafüggését.
Megállapítottuk a CpCo(CO)2, CpCo(CO)2+, CpCoCO+ és a CpCo+ gázfázisú képzõdéshõjét.
A CpCo(CO)2-hoz hasonlóan TPEPICO-vizsgálatokat végeztünk a CpMn(CO)3 molekulára.ICH2CNKvantumkémiai számításokat végeztünk a CpMn(CO)3 molekulára, valamint a CpMn(CO)3+, CpMn(CO)2+ CpMnCO+ és a CpMn+ ionokra, megvizsgáltuk a geometria disszociáció során bekövetkezõ változásait.
Megvizsgáltuk az átmeneti állapotok helyét a variációs átmeneti állapot elmélet alapján.
Elvégeztük a kobalt-komplexnél már említett RRKM-számításokat, letörési görbe, és TOF-tömegspektrum-modellezést erre a vegyületre is, meghatároztuk a konszekutív karbonil-disszociációk energetikáját, valamint kinetikáját.
A kísérleti adatok alapján megállapítottuk a CpMn(CO)3+, a CpMn(CO)2+, a CpMnCO+ és a CpMn+ gázfázisú képzõdéshõjét.
Kvantumkémiai módszerrel kétféle megközelítéssel kiszámítottuk a disszociációk során keletkezõ ionok képzõdéshõit; megállapítottuk, hogy az adiabatikus felület feltételezése vezet a kísérlettel legjobban egyezõ eredményekre.
A kísérleti és számított energetikai adatok kombinálásával becslést adtunk a semleges CpMn(CO)3 molekula, és a belõle keletkezõ fragmensek MnCO kötési energiájára.
A fentieken túl ennél a molekulánál sikerrel modelleztünk párhuzamos disszociációs kinetikát, ezáltal meghatároztuk a két párhuzamos reakció (I-, illetve CH2CN-vesztés) energetikáját, és sebességét.Köszönetet mondok Dr. Szepes László professzor úrnak, hogy munkámat kutatócsoportjában lehetõvé tette és tanácsaival segítette. Szintén hálával tartozom Thomas Baer professzornak a TPEPICO spektroszkópiai együttmûködésért.Az irodalmi adatokból a negatív ion körfolyamat alapján megállapítottuk a ·CH2CN-gyök, valamint ennek általunk mért az irodalmival jól egyezõ ionizációs energiája segítségével a CH2CN+-ion kísérleti képzõdéshõjét.
A TPEPICO-megjelenési energiák alapján meghatároztuk a jódacetonitril eddigi legpontosabb képzõdéshõjét, mely az eddigi számított értékeknél mintegy 20 kJ/mol-lal magasabbnak adódott.
Ionized States and the Dissociation of Organometallic Compounds
Ph.D. thesis
Eötvös Loránd University, Budapest
2000
Abstract
The present work focuses on three topics. First, lone-pair interactions in tetramethyldiarsane and tetramethyldistibane are investigated using ultraviolet photoelectron spectroscopy (UPS) and quantum-chemical methods. The second subject is the bonding mode of the tetrahydridoborato ligands in a zirconium complex. Third, threshold photoelectron photoion coincidence spectroscopic (TPEPICO) results of transition metal carbonyl complexes are presented.
The He(I) photoelectron spectrum of tetramethyldistibane was recorded.
These measurements gave clear evidence of the existence of the gauche
and the
anti conformer in the gas phase. The overall structure of
the spectrum is similar to that of tetramethyldiarsane and -diphosphane.
Systematic theoretical investigations of the arsenic and antimony compounds
raised questions concerning the original interpretation of the spectra.
Full potential energy curves along the C-E-E-C torsion (E=As, Sb) were
calculated at the Hartree-Fock (HF) level and complete geometry optimizations
were performed at HF, second order Many-Body Perturbation Theory (MBPT(2))
and Coupled-Cluster Singles and Doubles (CCSD) levels of theory. Two conformers,
anti
and gauche, have been found in accordance
with the experimental observations. The calculated geometries are in
good agreement with the electron diffraction results. Ionization energies
were computed by the equation-of-motion coupled-cluster (EOM-CC) method.
The calculations predict a substantial lone-pair splitting for both conformers
which contradicts the original assignment of the photoelectron spectrum
of tetramethyldiarsane by Cowley et al. According to the new assignment,
the first and third bands belong to the lone pairs of the anti rotamer,
the second band is attributed to the n+ lone-pair combination
in the gauche conformer, while the peak of the n combination
is merged with the intense first band previously ascribed exclusively to
the anti rotamer. The photoelectron spectrum of tetramethyl distibane
is shown in Figure 1.
Figure 1. The photoelectron spectrum of Me4Sb2
The conformer ratios of the two conformers of the arsenic compound calculated from the present assignment are in good agreement with the quantum-chemical results. Since our assignment contradicts Dewars theory, which the original assignment was based on, a new explanation is presented. We find that the s-character of the lone-pair orbitals increases in the order of nitrogen, phosphorus, arsenic and antimony which explains not only the increasing pyramidalization of the Me2E moiety but also the increasing splitting of the energy of the lone-pair MOs.
The geometry and the electronic structure of bis(tetrahydridoborato) bis(cyclopentadienyl) zirconium(IV) was investigated by UPS, X-ray diffraction and ab initio quantum-chemical methods (Fig. 2.). The photoelectron spectrum was recorded both at He(I) and He(II) photon energies, which was assigned on the basis of the quantum-chemical calculations and comparison with analogous complexes. Full geometry optimization was performed at the HF and at the MBPT(2) levels of theory. The obtained geometry is in good agreement with the anticipated bidentate ligation of the BH4 groups. The measured Zr-B distance from the X-ray studies gives further evidence for the abovementioned bonding mode of the tetrahydridoborato ligands. The ionization energies of the complex were calculated using the Outer Valence Greens Function (OVGF) method.
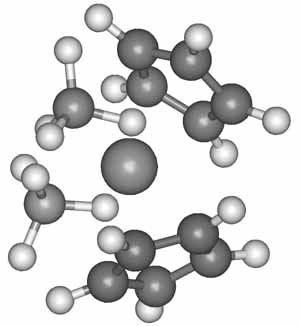
Figure 2. The calculated structure of Cp2Zr(BH4)2
TPEPICO spectroscopy has been used to investigate the dissociation dynamics of cyclopentadienyl cobalt dicarbonyl ion, CpCo(CO)2+ (Fig. 3.). The dissociation proceeds by the sequential loss of the two CO molecules. Both reactions proceed with no reverse activation energies and are slow near their dissociation onset. The 0K onset was determined by modeling the measured dissociation rate constants with RRKM theory utilizing the variational transition state theory to locate the transition states. The ionization energy of CpCo(CO)2+ was measured from the threshold photoelectron spectrum to be 7.35 eV. The neutral CpCo(CO)2+ gas phase heat of formation of -117±10 kJ/mol was determined from the liquid value by measuring the heat of vaporization of 52.12±0.68 kJ/mol.
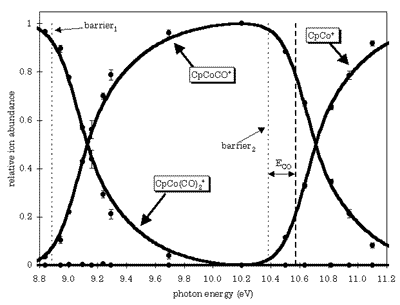
Figure 3. The breakdown curve of CpCo(CO)2
The first Co-CO bond energy in the CpCo(CO)2+ was found to be 1.54±0.01 eV (148.8±1 kJ/mol) giving a (298K) gas phase heat of formation of 858±10 kJ/mol for CpCoCO+. The second Co-CO bond energy in CpCoCO+ was measured to be 1.49±0.01 eV (144.0±1 kJ/mol) resulting in a (298K) gas phase heat of formation of CpCo+ to be 1113±10 kJ/mol.
TPEPICO studies of a similar complex, cyclopentadienyl manganese tricarbonyl, CpMn(CO)3 was also carried out.. The ionization energy of CpMn(CO)3 was measured from the threshold photoelectron spectrum to be 7.69 ± 0.01 eV. The dissociation of the CpMn(CO)3+ ion proceeds by the sequential loss of three CO molecules. The first and third CO loss reactions were observed to be slow (lifetimes in the m s range). By simulating the resulting asymmetric time-of-flight peak shapes and breakdown diagram, 0 K onsets for three daughter ions were determined to be 8.80 ± 0.01, 9.43 ± 0.02 and 10.51 ± 0.03 eV, respectively. The three successive Mn-CO bond energies in the CpMn(CO)3+ were found to be alternating with values of 1.11 ± 0.01 eV, 0.63 ± 0.02 eV and 1.08 ± 0.03 eV, respectively. The 298 K gas phase heats of formation of CpMn(CO)3+, CpMn(CO)2+, CpMnCO+ and CpMn+ are obtained to be 262 ± 9, 483 ± 9, 656 ± 9 and 875 ± 10 kJ/mol, respectively. Using the known heats of formation of Cp and Mn+, the Cp-Mn+ bond energy was also found to be 3.75 ± 0.12 eV. By scaling theoretical calculated neutral bond energies with the experimental information derived in this study, the successive Mn-CO bond energies were estimated to be 2.07, 1.21 and 1.14 eV, respectively, while the Cp-Mn bond energy was found to be 2.39 eV. Comparison between the quantum chemical calculations and experimental values shows that the loss of CO groups follows an adiabatic path, in which electronic spin on the metal center is not conserved.
Because of interesting problems in the modelling of the spectroscopic results, a strictly organic compound, iodoacetonitrile (ICH2CN) has also been investigated by TPEPICO spectroscopy. The 0K onsets for the following products were determined: +CH2CN + I· (12.188 ± 0.005 eV) and ·CH2CN + I+ (12.345 ± 0.010 eV). From the difference between these two values the ionization energy of the ·CH2CN was found to be 10.294 ± 0.010 eV. By using a thermodynamic cycle that involves the gas phase acidity of CH3CN, the electron affinity of the ·CH2CN radical, and an accurate heat of formation of acetonitrile, a ÄfHo298(ICH2CN) of 172.5 ± 4.0 kJ mol-1 is derived. This latter value is considerably higher than the best theoretical value of 153 kJ mol-1.
The authors gratefully acknowledge the financial support of the Department
of Energy (US), the ELTE Peregrinatio Foundation, the Department
of Education (Hungary), the Hungarian National Research Fund,
the Pro Scientia Foundation, the Soros Foundation and the
Foundation
for Chemical Education (ELTE).
Publications
The dissociation kinetics of energy selected CpMn(CO)3+
ions studied by threshold photoelectron-photoion coincidence spectroscopy,
Li, Y.; Sztáray, B.; Baer, T. Journal of the American
Chemical Society, accepted
The dissociation dynamics and thermochemistry of energy selected CpCo(CO)2+
ions, Sztáray, B.; Baer, T. Journal of the American Chemical
Society, 122 (2000) 9219
A photoelectron and photoion coincidence study of the ICH2CN
dissociation: Thermochemistry of CH2CN, +CH2CN,
and ICH2CN, Lafleur, R.D.; Sztáray, B.; Baer, T.
Journal of Physical Chemistry A, 104 (7) (2000) 1450
The geometry and electronic structure of bis(cyclopentadienyl)-bis(tetrahydridoborato)-zirconium(IV),
Sztáray, B.; Rosta, E.; Böcskey, Zs.; Szepes, L. Journal
of Organometallic Chemistry, 582 (1999) 267
Structure and photoelectron spectrum of tetramethyldiarsane, Sztáray,
B.; Szalay, P. Journal of the American Chemical Society, 119
(1997) 11926
Rotational isomerism in tetramethyldistibane studied by UV photoelectron-spectroscopy,
Sztáray, B.; Nagy, A.; Szepes, L.; Breunig, H.J. Journal of Organometallic
Chemistry, 515 (1996) 249
| Vissza a tartalomjegyzékhez
Back to Contents |
http://www.kfki.hu/chemonet/
http://www.chemonet.hu/ |