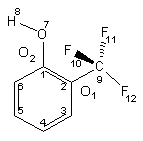
1a : Q1~60o, Q2=180o (minimum)
1b : Q1=0o, Q2=0o (nyeregpont)
1c : Q1~60o, Q2=0o (nyeregpont)
1d : 0o<Q1<60o, Q2<0o (minimum)
Kovács Attila
Kvantumkémiai számítások néhány alkalmazása a szerkezetkutatásban
Budapesti Mûszaki Egyetem
1997
Bevezetés
A számítástechnika rohamos fejlõdésével az utóbbi években a kvantumkémiai számítások kezdenek tért hódítani a nagyobb méretû molekulák körében is. A kvantumkémiai módszerek egyik leggyakoribb alkalmazása a rokon vegyületek vizsgálata, ahol nem annyira a meghatározott tulajdonságok különbözõ hibákkal terhelt abszolút, hanem inkább relatív értékei a lényegesek. A szerkezetkutatásban fontos alkalmazási területek a konformációs analízis, rotációs gát meghatározás, hasonló molekulák szerkezeti különbségeinek vizsgálata, rezgési analízis. Jelen értekezésben a CF3 és OH csoportok közötti intramolekuláris hidrogénkötés vizsgálatát, az N,N-dimetil-karbamoil-klorid és néhány szervetlen molekula (tellúr-kloridok és lantán-halogenidek) rezgési analízisét mutatom be.
A CF3- és OH-csoportok közötti intramolekuláris hidrogénkötés vizsgálata
1. 2-trifluormetil-fenol [1]
A molekula jellegzetes konformerei a következõ torziós szögekkel jellemezhetõk (Q1=Ð F10C9C2C1; Q2== Ð H8O7C1C2):
|
1. ábra
1a : Q1~60o, Q2=180o (minimum) 1b : Q1=0o, Q2=0o (nyeregpont) 1c : Q1~60o, Q2=0o (nyeregpont) 1d : 0o<Q1<60o, Q2<0o (minimum) |
A konformerek relatív stabilitását különbözõ kvantumkémiai szinteken (HF, MP2, különbözõ DFT módszerek) vizsgáltuk. Valamennyi számítás a hidrogénkötéses syn típusú szerkezeteket mutatja stabilabbnak az anti 1a-val szemben, a globális minimum 1-3 kcal/mol-al kedvezõbb energetikailag. A számítások általában az 1d geometriát jelzik a globális minimumnak (a várt 1b-vel szemben), amelyben mind az OH hidrogén, mind a vele kölcsönhatásban levõ fluor kitér a benzolgyûrû síkjából.
A Bader-féle topológiai analízist alkalmazva az 1d konformerben kötés kritikus pontot kaptunk a hidrogénkötésben résztvevõ fluor és OH hidrogén között. A Laplace-koncentráció és az elektronsûrûség kötés kritikus pontbeli értéke a hidrogénkötés erõsen ionos jellegére utal.
A hidrogénkötés hatása a molekula geometriájára a következõképpen összegezhetõ (referenciaként a fenol és trifluormetil-benzol szerepelt):
a hidrogénkötésben résztvevõ fluor CF kötése jelentõsen megnyúlik;
az OH kötés hossza gyakorlatilag nem változik, ellentétben a más vegyületeknél tapasztaltakkal
a CO és CCF3 kötések megrövidülnek;
a benzol gyûrûben a C1C2 kötés megnyúlik, a C3C4 és C5C6 megrövidül;
a kötésszögek közül a OC1C2 és a COH szög nõ jelentõsebben.
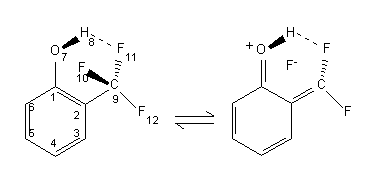
A fenti geometriai változások az alábbi ábrán vázolt rezonáns határszerkezettel jellemezhetõ irányú elektronsûrûség átrendezõdésre utalnak (2. ábra):
2. 2-trifluormetil-rezorcin és bisz-2,6-trifluormetil-fenol [2]
A 2-trifluormetil-rezorcin konformációs analízisénél a syn-syn rotamer környezetében szintén egy nagyon lapos potenciális energia felületet találtunk. A számítások során kapott globális minimum aszimmetrikus, két különbözõ F...HO kölcsönhatást tartalmaz. A Cs szimmetriájú, két ekvivalens F...HO hidrogénkötést tartalmazó geometria egy alacsonyan fevõ nyeregpont (0.02 kJ/mol az MP2/6-31G** szinten). A 2,6-bisz(trifluormetil)-fenol szerkezete hasonló a 2-trifluormetil-fenoléhoz.
A geometriai paraméterek elemzése alapján a következõ megállapításokat tehetjük:
a 2,6-bisz(trifluormetil)-fenolban a második CF3-csoport jelentéktelen befolyással van az intramolekuláris hidrogénkötésre;
a 2-trifluormetil-rezorcinban a F...HO hidrogénkötés kis mértékben erõsebb, mint a 2-trifluormetil-fenolban és 2,6-bisz(trifluormetil)-fenolban;
az OH kötéshossz gyakorlatilag nem változik;
az intramolekuláris hidrogénkötés eredményeként a 2-tri-fluormetil-fenolnál bemutatott irányú elektronsûrûség átrendezõdés történik.
3. 2-trifluormetil-vinil-alkohol [3]
A molekulában levõ F...HO hidrogénkötést a 2-trifluor-metil-fenoléval összevetve vizsgáltuk. A konformációs tulajdonságok vizsgálatánál egy lényeges különbséget találtunk a két rendszer között: míg a 2-trifluormetil-fenol globális minimumában mind az OH csoport, mind a vele kölcsönható fluor jelentõsen kitér a benzolgyûrû síkjából, addig a 2-tri-fluormetil-vinil-alkoholban a hidrogénkötéses hattagú gyûrû teljesen sík. Ez a különbség egyértelmûen a benzol gyûrû hatására vezethetõ vissza. Úgy tûnik, a hidrogénkötés a 2-tri-fluormetil-fenolban túl gyenge ahhoz, hogy kompenzálni tudja a nem-planáris elrendezõdést preferáló elektronikus és sztérikus hatásokat.
A fenti modellvegyületekben tapasztaltak alapján a CF3 és OH csoportok közötti intramolekuláris hidrogénkötésrõl a következõ általános megállapításokat tehetjük:
Ezen intramolekuláris F...HO hidrogénkötések gyengék, sokkal gyengébbek mint a hasonló O...HO és N...HO-származékokban találtak.
A hidrogénkötések geometriai következményeit illetõen a jelentõsebb effektusok a CF3 csoportban vannak, eltérõen más O...HO és N...HO rendszerektõl, ahol a tapasztalat az OH kötés számottevõ megnyúlását is mutatja.
A hidrogénkötések geometriai következményei egy rezonáns határszerkezetekkel jellemezhetõ tendenciájú elektronsûrûség átrendezõdésre utalnak.
Az N,N-dimetil-karbamoil-klorid rezgési analízise [4]
Elsõként végeztük el a molekula rezgési spektroszkópiai vizsgálatát (gáz és folyadékfázisú IR spektrumok alapján). Az IR spektrumok értelmezésére a "skálázott kvantum-mechanikai" (SQM) módszert használtuk, a molekula B3-LYP/6-31G* harmonikus rezgési erõterét empírikusan skálázva. Az empírikus korrekcióhoz Rauhut és Pulay ezen szintû kvantumkémiai számításokhoz meghatározott skála-faktorait használtuk. Mivel a klóratom deformációs rezgéseire az irodalomban nem találtunk skálafaktorokat, ezeket mi határoztuk meg. Modellvegyületként az acetil-kloridot választottuk, melynek rezgési spektruma az irodalomból ismert.
Az N,N-dimetil-karbamoil-klorid 30 normálrezgésébõl 22-t azonosítottunk az IR spektrumokban. A kísérleti és SQM frekvenciák közötti eltérés (súlyozott átlagos deviáció, ahol súlyként a kísérleti frekvenciák reciproka szerepelt) 10.0 cm-1 volt. A normálrezgések sávjain kívül két felhangot és egy kombinációs sávot azonosítottunk a 2500-1000 cm-1 tartományban.
Szervetlen halogenidek rezgési analízise
1. Tellúr-kloridok [5,6]
A tellúr-tetraklorid gázfázisú IR spektrumát az 1000-25 cm-1-es tartományban 425 és 675 K hõmérsékleteken készítettük el. Az MP2 módszer és Hay és Wadt relativisztikus törzspotenciáljainak alkalmazásával (Te) meghatároztuk a molekula harmonikus rezgési erõterét. A "skálázott kvantummechanikai (SQM)" módszer segítségével elvégeztük a molekula teljes rezgési analízisét, melynek eredményeképpen tisztáztuk a kétséges B2 deformációs rezgést, azonosítottuk az IR spektrumunkban a B1 deformációs rezgést, és megbízható értéket határoztunk meg az IR inaktív A2 rezgés frekvenciáját illetõen. A skálázott rezgési erõteret a molekula elektrondiffrakciós szerkezetanalíziséhez használtuk fel.
A tellúr-diklorid példáján vizsgáltuk, hogy a TeCl4 mo-lekulára meghatározott skálafaktorok mennyire vihetõk át hasonló származékokra. A TeCl2 a priori frekvenciái nagyon jól egyeztek az eddig ismert két kísérleti adattal (maximális eltérés 4 cm-1).
2. Lantán-halogenidek gázfázisú IR spektruma [7]
A LaCl3, LaBr3 és LaI3 gázfázisú IR spektrumát az 1000-25 cm-1-es tartományban 1400-1450 K-en vettük fel. A spektrumokban két széles - általában komplex szerkezetû - sáv látható, egy az LaX (X=halogén) vegyértékrezgési, egy pedig a deformációs rezgések tartományában. A kísérleti spektrumokat kvantumkémiai számítások (MP2 módszer és Hay-Wadt törzspotenciálok) és sávkontúr analízis segítségével értelmeztük. Megállapítottuk, hogy a spektrumokban a molekulák négy normálrezgése közül kettõ (n2 és n3) ad intenzív elnyelési sávot. A n1 normálrezgés (a geometria piramisos illetve planáris jellegétõl függetlenül) egyáltalán nem várható az IR spektrumokban, a kisebb intenzitású n4-et pedig n2 R ága takarja.
Selected applications of quantum chemical calculations in structural chemistry
Ph.D. thesis
Technical University of Budapest
1997
Introduction
Due to the revolutionary development of computer techniques the application of the quantum chemical calculations for larger molecules is increased in the last years. One of the most popular applications of these computations is the investigation of similar compounds, where the relative values of the calculated parameters are more characteristic than the absolute values influenced by different errors. The most important applications in structural chemistry are the conformational analysis, determination of rotational barrier, vibrational analysis, study of structural differences in related compounds. In the present thesis I present my recent results about the intramolecular hydrogen bonding between CF3 and OH groups, and the vibrational analysis of some inorganic halides (tellurium chlorides and lanthanum trihalides).
Investigation of the intramolecular hydrogen bonding interaction between CF3 and OH groups
1. 2-Trifluoromethylphenol [1]
The most important conformers of the molecule may be characterised by the following (Q1=Ð F10C9C2C1; Q2== Ð H8O7C1C2, see Scheme 1 in the Hungarian text):
1a : Q1~60o, Q2=180o (minimum)
1b : Q1=0o, Q2=0o (nyeregpont)
1c : Q1~60o, Q2=0o (nyeregpont)
1d : 0o<Q1<60o, Q2<0o (minimum)
The relative stability of the conformers was investigated at different quantum chemical levels (HF, MP2, DFT). According to the computations the hydrogen-bonded syn geometries are more stable with respect to the anti 1a, the global minimum being favoured by 1-3 kcal/mol. In contrary to the expected 1b we found the 1d geometry as the global minimum, where both the OH hydrogen and the fluorine interacting with it are out of the plane of the benzene ring.
In 1d we found a bond critical point in Bader's sense between the OH hydrogen and the fluorine participating in the hydrogen bond. The electron density and the Laplacian at the bond critical point are in agreement with the strong ionic character of the present hydrogen bonding interaction.
The effect of hydrogen bonding on the molecular geo-metry is the following (as reference we used the computed geometries of phenol and trifluoromethyl-benzene):
the CF bond participating in the hydrogen bonding is lengthened
the OH bond length is practically unchanged (in contrary to the general observation for hydrogen bonding)
the CO and CCF3 bonds are shortened
the C1C2 bond of the benzene ring is lengthened while the C3C4 and C5C6 bonds are shortened
among the bond angles OC1C2 and COH are opened
All the geometrical changes indicate an electron density redistribution which may be characterised by the resonance structure shown in Scheme 2 (Hungarian text).
2. 2-Trifluoromethylresorcinol and bis(2,6-trifluoromethyl)-phenol [2]
The conformational analysis of 2-trifluoromethyl-resor-cinol resulted in a very flat potential energy valley around the syn-syn rotamer. The global minimum is asymmetric, contains two different F...H(O) interaction. The geometry with two equivalent F...H(O) interactions (Cs symmetry) is a low-lying saddle-point (by 0.02 kJ/mol at the MP2/6-31G** level). The structure of bis(2,6-trifluoro-methyl)phenol is similar to that of 2-trifluoromethylphenol.
From the analysis of the geometrical parameters our conclusions are the following:
the second CF3 group in bis(2,6-trifluoromethyl)phenol has negligible influence on the intramolecular hydrogen bond
the F...H(O) hydrogen bond in 2-trifluoromethyl-resor-cinol is somewhat stronger than that in bis(2,6-trifluoro-methyl)phenol and in 2-trifluoromethylphenol
the OH bond length is practically unchanged
the hydrogen bonding interaction results in an electron density redistribution similar to that found in 2-tri-fluoromethylphenol
3. 2-Trifluoromethylvinyl alcohol [3]
The F...H(O) hydrogen bond in the molecule was compared with that in 2-trifluoromethylphenol. While in the global minimum of 2-trifluoromethylphenol both the OH hydrogen and the fluorine are out of the plane of the ring, in 2-trifluoromethylvinyl alcohol the hydrogen-bonded six-ring is planar. This difference may be attributed to the effect of the benzene ring. It seems, that the hydrogen bonding interaction in 2-trifluoromethylphenol is too weak and cannot compensate the different electronic and steric effects favouring a non-planar arrangement.
Our general conclusions for the intramolecular hydrogen bonding interaction between the CF3 and OH groups are the following:
these intramolecular F...H hydrogen bonds are weak, much weaker than those found in similar O...HO and N...HO derivatives.
the geometrical consequences of hydrogen bonding are more pronounced in the CF3 group, in contrary to similar O...HO and N...HO systems, where in general a considerable lengthening of the OH bond was found
the geometrical consequences of hydrogen bonding indi-cate an electron density redistribution which may be characterised by resonance structures
Vibrational analysis of N,N-dimethylcarbamyl chloride [4]
The molecule was investigated by vibrational spectroscopy in the present study for the first time. The FT-IR spectra of the gas and liquid were interpreted using the "scaled quantum mechanical" (SQM) method based on the B3-LYP/6-31G* computed harmonic force field. For scaling we used the scale factors developed recently by Rauhut and Pulay. We could not find any scale factor for the deformation vibrations of chlorine in the literature, hence we developed this scale factor based on the known vibrational spectra of acetyl chloride.
From the 30 normal vibrations of N,N-dimethyl-carbamyl chloride 22 were identified in the vibrational spectra. The weighted mean deviation between the experimental and SQM frequencies (where weighting was done by the inverse of the experimental frequency) was 10.0 cm-1. Besides the fundamentals we assigned two overtones and a combination band in the range of 2500-1000 cm-1.
Vibrational analysis of inorganic halides
1. Tellurium chlorides [5,6]
We recorded the gas-phase IR spectra of TeCl4 in the 1000-25 cm-1 range at 425 and 675 K. The harmonic force field of the molecule was calculated at the MP2 level using the relativistic effective core potentials of Hay and Wadt for tellurium. We performed the complete vibrational analysis of the molecule using the SQM method. In the course of this work the discrepancy concerning the B2 deformation mode was cleared, the B1 deformation mode was assigned and a reliable SQM frequency was obtained for the A2 mode inactive in the infrared. The scaled force field was further used at the electron diffraction structure analysis of the molecule.
We investigated the transferability of the scale factors developed on TeCl4. Applying these scale factors for the computed force field of TeCl2 the a priori TeCl2 frequencies are in very good agreement with the available experimental results (maximal deviation 4 cm-1).
2. Gas-phase FT-IR spectra of lanthanum trihalides [7]
The gas-phase FT-IR spectra of LaCl3, LaBr3 and LaI3 were recorded in the 1000-25 cm-1 range at 1400-1450 K. In general two broad complex bands are seen in the spectra, one in the stretching and one in the bending range. The experimental spectra were interpreted using quantum che-mical calculations (MP2 method and the relativistic effective potentials of Hay and Wadt). According to our present study the n3 and n2 fundamentals of the trihalides can be assigned to the observed bands. The n1 mode (independent from the pyramidal or planar character of the compounds) cannot be expected to observe in the infrared spectra, while the low intense n4 is hidden by the R branch of n2.
References
1. A. Kovács, I. Kolossváry, G. I. Csonka, I. Hargittai: Theoretical study of intramolecular hydrogen bonding and molecular geometry of 2-trifluoromethylphenol. Journal of Computational Chemistry, 17 (1996) 1804.
2. A. Kovács, I. Hargittai: Hydrogen Bonding in 2-Trifluo-romethylresorcinol and 2,6-Bis(trifluoromethyl)-phenol and Its Geometrical Consequences. J. Mol. Struct., in press
3. A. Kovács, I. Hargittai: Hydrogen-bonding interactions of the trifluoromethyl group: 2-trifluoromethyl-vinyl alcohol. Int. J. Quantum Chem. 62 (1997) 645.
4. A. Kovács, V. Izvekov: FT-IR investigation of N,N-dimethylcarbamyl chloride. J. Mol. Struct., 410-411 (1997) 403.
5. A. Kovács, K.-G. Martinsen, R.J.M. Konings: The gas-phase vibrational spectrum and molecular geometry of TeCl4. J. Chem. Soc., Dalton Trans., (1997) 1037.
6. A. Kovács, R.J.M. Konings: Vibrational spectra of TeCl4 and TeCl2. J. Mol. Struct., 410-411 (1997) 407.
7. A. Kovács, A.S. Booij, R.J.M. Konings: High-temperature infrared spectra of LaCl3, LaBr3 and LaI3 Chemical Physics Letters, 268 (1997) 207.
| Vissza a tartalomjegyzékhez Back to Contents |
http://www.kfki.hu/chemonet/ http://www.ch.bme.hu/chemonet/ |