Beszélgetés Keserû György Miklóssal
A 2001. évi Oláh György-díjat a szakalapítvány
kuratóriuma Keserû György Miklósnak ítélte
oda. A kitüntetett 1991-ben végzett a Budapest Mûszaki
Egyetemen, 1993-ban kandidátusi fokozatot szerzett. A Richter Gedeon
Rt. Molekulatervezési Osztályát vezeti, a Mûegyetem
Kémiai Informatikai Tanszékén meghívott kutató.
Beszélgetésünkre a tanszéken került sor.
Nemrég fejezõdött be az órája. Mi volt a téma?
 A
Molekulatervezés címû tantárgyat tanítom.
Ez kötelezõen választható tárgy, általában
nyolc-tíz-tizenkét hallgató veszi fel egy félévben.
A számítógépes molekulatervezés elméletének,
illetve néhány gyakorlati alkalmazásának jut
hely egy féléves tantárgy keretei között.
Gyakran jönnek hallgatók az ELTE-rõl és
doktoranduszok is hallgatják az órákat.
A
Molekulatervezés címû tantárgyat tanítom.
Ez kötelezõen választható tárgy, általában
nyolc-tíz-tizenkét hallgató veszi fel egy félévben.
A számítógépes molekulatervezés elméletének,
illetve néhány gyakorlati alkalmazásának jut
hely egy féléves tantárgy keretei között.
Gyakran jönnek hallgatók az ELTE-rõl és
doktoranduszok is hallgatják az órákat.
Molekulamechanikai vagy ab initio módszerekkel foglalkoznak?
Alapvetõen a molekulamechanika és a konformációs analízis témakörébõl merítünk. Nagy hangsúlyt fektetünk a biomolekulák modellezésére, amelyek a gyógyszertervezésben is kiemelkedõ jelentõségûek, ezért elsõsorban olyan számítási módszereket alkalmazunk, amelyek a biomolekuláris problémákat is kezelik.
Az elõadás vagy a gyakorlat felel meg jobban meg a témának?
A félévet elõadással vagy inkább szemináriummal kezdjük, amely késõbb, amikor az alapismereteket sikerült összegyûjteni, gyakorlattá alakul. Ez azt jelenti, hogy itt a Kémiai Informatikai Tanszéken rendelkezésre álló szoftver és hardver háttér mellett alkalmazhatják a hallgatók a frissen szerzett ismereteket. Ebben jelentõs szerepet játszik, hogy nagyon jó kapcsolatunk van a TRIPOS-szal, amely a szoftvereinknek egy részét adja, és a Schrödinger nevû amerikai szoftvercég is együttmûködik velünk.
Egy nagyon szélsõséges álláspont szerint a molekulamechanikai modellek igazából arra jók, hogy a cégek jelentéseiben szép színes térbeli ábrák készülhessenek.
Általában a kvantumkémikusok képviselik ezt az álláspontot. Azok a piacra került gyógyszerek cáfolják meg alapvetõen az említett felfogást, amelyek elõállításához már a kutatás kezdeti szakaszában számítógépes molekulatervezési eljárásokat használtak.
A módszert az 1970-es években kezdték alkalmazni, és a statisztikai megközelítésen alapuló szerkezet-hatás összefüggések területén hozott elõször gazdasági sikereket. A Captopryl nevû vérnyomáscsökkentõ volt az elsõ olyan gyógyszer, amelynél tudatosan alkalmaztak ilyen eljárást. Ezt követõen számos QSAR-alapú alkalmazásra került sor.
A szerkezeti biológia kiteljesedésével újra kiemelkedõ jelentõséget kaptak ezek a módszerek, mert a nagy felbontású kísérleti röntgen- vagy NMR-fehérjeszerkezetek képezik a számítás alapját. Ez nagymértékben megváltoztatja a pontosságról korábban alkotott képet, hiszen a biológiai szerkezetet nem ab initio módon "kitaláljuk", hanem egy kísérleti szerkezet környezetében kezdünk el dolgozni. A szerkezeti biokémia a molekulatervezéssel együttmûködve olyan HIV-proteáz inhibitorok megjelenéséhez vezetett a gyógyszerkutatásban, amelyek már piacra is kerültek.
Visszatérve a felvetett gondolatra: igyekeznünk kell, hogy ne szélsõséges megközelítések mentén vizsgáljunk egy tudományterületet. Próbáljunk meg tárgyilagosak maradni, ami ebben az esetben azt jelenti: tudom, hogy egy adott megközelítés érvényessége hol kezdõdik és hol ér véget.
Úgy látom, hogy a gyógyszerkutatásban nincsenek egyedül üdvözítõ utak. A nyolcvanas évek közepéig sokan azt hitték, hogy a racionális gyógyszertervezéssel a gyógyszerkutatás minden problémáját meg lehet oldani. Késõbb természetesen kiderült, hogy ez nem így van. A kombinatorikus kémiáról is tudjuk már, hogy csak bizonyos keretek között alkalmazható sikerrel. A nagy áteresztõképességû tesztelés is ilyen. Érdekes ugyanakkor, hogy a kvantumkémia elég elenyészõ szerepet kap a gyógyszerkutatásban. Ennek elsõsorban méretbeli okai vannak. Természetesen mi is használunk megfelelõ szinten kvantumkémiai számításokat, amikor kis molekulákat vizsgálunk és potenciális gyógyszerekrõl szeretnénk információt kapni.
A geometriát vagy a töltéseloszlást vizsgálják kvantumkémiai módszerekkel?
Mindkettõt. Elvi okok miatt nem utasítunk el semmilyen módszert azért, mert azt valaki más használja; egyetlen kritériumunk van, a hasznosság.
Azzal is tartozunk az igazságnak, hogy az erõterek, a parametrizálások olyan jók, hogy a legtöbb molekulamechanikai módszer a kis molekulákra majdnem az ab initio szerkezetet adja.
A parametrizálásnak ez az útja valóban rendkívül hatékony. Több olyan erõtér is létezik, ahol már nem is választottak kísérleti megközelítést, csak ab initio parametrizálást. Ez visszavezethetõ arra a problémára is, hogy a röntgenszerkezeteket a szilárd fázisban határozzák meg, de gázfázisban számolunk, vagy a legjobb esetben valamilyen folyadékszimulációval igyekszünk megközelíteni a valóságot. Azért célszerû ab initio számításokat használni molekulamechanikai erõterek parametrizálására, mert ilyenkor nincs fázisprobléma, mindkét számítás a gázfázisra vonatkozik.
Úgy érzem, hogy a kvantumkémikusok is egyre inkább elmozdulnak a biológiai rendszerek irányába. Mi jobban otthon vagyunk ezen a területen, viszont segítségre szorulunk egy-két olyan kérdésben, amelyben õk specializálódtak és tudást szereztek.
Miben segíthetnek most a kvantumkémikusok?
A kismolekulás töltésszámításokban, illetve különbözõ biokémiai mechanizmusoknak a megértésében számíthatunk az õ segítségükre. Ha valamilyen biokémiai mechanizmusba akarunk beleszólni egy új gyógyszerjelölttel, célszerû elõbb a mechanizmust megismerni, hogy tudjuk, mi az, amit befolyásolni szeretnénk. A sûrûségfunkcionál módszerek megjelenése ezen a területen áttörést hozott, mert ezekkel a módszerekkel lényegesen kedvezõbb számítási teljesítmény mellett szerezhetünk információt akár kiterjedtebb rendszerekrõl is. Ezek a módszerek számos esetben segíthetnek egy biokémiai mechanizmus feltárásában.
A sûrûségfunkcionál módszert mi is alkalmaztuk a közelmúltban egy olasz csoporttal együttmûködve a HIV-integráz fehérje mûködésének modellezésére. Ez a fehérje a vírus genetikai állományát integrálja a gazdaszervezethez. Ettõl kezdve a gazdaszervezet állítja elõ a vírus genomját. Mi az integrálási lépést vizsgáltuk sûrûségfunkcionál módszerek alkalmazásával.
Ugyancsak fontosak a QMMM számítások, ahol a QM a kvantummechanikát, az MM a molekulamechanikát jelenti. Ezek kettõs megközelítések, amikor arról van szó, hogy egy fehérje reakciócentrumát, ahol a kémiai átalakulás zajlik, kvantumkémiával modellezzük annak érdekében, hogy megfelelõ reakcióút-számításokat tudjunk végezni, a reakcióúton az átmeneti állapotokat, köztitermékeket azonosítani lehessen, és ezáltal a mechanizmust meg tudjuk érteni. A reakciócentrumot körülvevõ, kiterjedt fehérjeburkot azonban molekulamechanikával számítjuk. A QMMM számítások tehát a kooperatív erõfeszítések jó példái.
Annál is inkább, mert a molekulamechanikai programok, például a SYBYL, állandó kötésekkel számolnak, míg az ab initio módszerek esetén csak atomokkal van dolgunk, és az a kérdés, hogyan rendeljünk hozzájuk kötéseket.
Igen, itt az a fontos, hogy addig, amíg azt akarjuk megvizsgálni, hogyan kötõdik egy kis molekula egy fehérjéhez, vagy milyen kötési lehetõségek adódnak, a molekulamechanikai alapmódszerek nagyon megbízhatóak. Amikor új kovalens kötések alakulnak ki, vagy meglevõ kovalens kötések szakadnak fel, egy szûkített tartományban a sûrûségfunkcionál módszerek vagy a kevert megközelítések, például a QMMM eljárások alkalmazhatók.
Az Oláh-díj átadásakor tartott elõadásában beszélt arról, hogy gyakran a szekvencia alapján kell meghatározni a térszerkezetet. Mi a tapasztalat, mennyire jók ezek a szerkezetmodellek?
Amikor az ember belekezd egy homológia-modellezési munkába, a szerkezettel való hasonlóság, illetve azonosság az alapvetõ szempont. Azt szokták mondani, hogy a hasonlóságnak 30% fölött kell lennie egy modellezett fehérje esetében. Ez valóban az alsó határ a hasonlósági modellezésben, nyilván annál nagyobb eséllyel kapunk jó szerkezetet, minél nagyobb a hasonlóság.
Az elõadáson bemutatott példáknak az volt a különlegességük, hogy nagyon stabil hasonlóságra épültek. Tudtuk, hogy a templátként használt és a modellezett fehérje ugyanabba a fehérjecsaládba tartozik, és tudtuk, hogy a feltekeredésük jellege is nagy valószínûséggel hasonló. Ennek a modellnek az elõállítása ezért nem okozott nagy szakmai kihívást. A feladat mindig akkor válik kritikussá, amikor az alacsony hasonlósági tartományban vagyunk, és ott próbálunk szerkezeti információt szerezni a fehérjérõl. Ha szoros analógiát tudunk, ezek a modellek nagyon jók lesznek, ezért nem volt meglepõ, hogy az illesztett röntgenszerkezet és a modellezett szerkezet esetében az összes nehéz atom helyzeténél 1,5, legfeljebb 2 angström eltérést kaptunk.
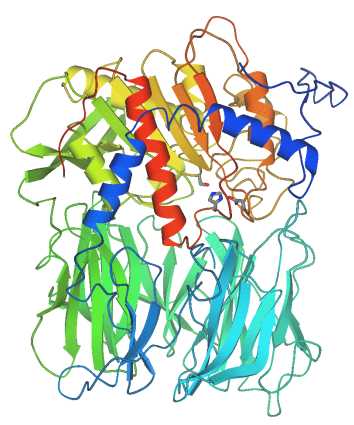
Az oligopeptidáz B homológiamodellezéssel prediktált szerkezete
Azok a szerkezetek, amelyek nagyon markáns hasonlóság
mellett készültek el, alkalmasak a kötõdés
energetikai viszonyainak kvalitatív vagy akár kvantitatív
vizsgálatára. Sajnos a biológiai vagy farmakológiai
szempontból legérdekesebb problémáknál
rendre nem áll fenn ez az eset, tehát az alacsony homológiatartományban
kell dolgoznunk. Ilyenkor két megközelítés van.
Készítünk például egy kezdeti modellt,
és ezt követõen, a szerkezetfinomítás
során speciális algoritmusokat alkalmazunk. A közelmúltban
Kolossváry Istvánnal egy egész fehérjére,
egy Jun-terminális kinázra hajtottunk végre konformációs
analízist. Ez azt jelenti, hogy ezzel az új módszerrel
több ezer szabadsági fok mentén tudunk konformációs
analízist végezni.
A JNK3 Jun-terminális kináz prediktált felcsavarodása
Más esetekben az a modell is alkalmazható, amelyiknek a pontossága korlátozott, mert alacsony volt a hasonlóság. Például egy vállalatnak van egy óriási vegyülettára, és arra szeretnénk választ kapni, hogy ennek az vegyülettárnak mely része az, amelyikbõl a legnagyobb valószínûséggel jöhet ki egy találat valamilyen tesztben. Elõször is készítünk egy durva modellt. A személetesség kedvéért idézzük fel a következõ játékot: van egy házikó, és ennek minden oldalán van egy geometriai alakzatot formáló ablak. A nyílásokba a megfelelõ formákat kell bedugni, például hengert, téglát, felhõt. A mi alacsony felbontású modellünk úgy mûködik, mint a házikó. A "háromszög alakú" zsebbe nem tudunk betenni egy "henger"-t. A "hengerek" tehát kiesnek és a molekula más részeire tudunk figyelni.
Mióta a humán genom ismert, van egy olyan elképzelés is, hogy a régi gyógyszertervezési utat meg kell fordítani.
Vannak kísérletek, amikor megfelelõ könyvtárakból szintetizálnak fehérjéket, és a kémiai proteomika módszerét használják: a fehérjéket "belelógatják" egy vegyületkeverékbe, ahol minden fehérje megtalálja a neki megfelelõ vegyületet. Ilyenkor tehát a vegyületekhez kötõdõ fehérjéket keresik. Nem tudni, mire jó egy-egy fehérje, de majd jó lesz valamire.
Mi az elméleti utat keressük. Véleményünk szerint akkor alkalmazható ez a megközelítés a molekulatervezésre sikerrel, ha a humán genom program által hordozott információból szerkezeti információt tudunk kinyerni. A kérdés az, hogyan juthatunk hozzá a szerkezeti információhoz. Nekünk fehérjeszekvenciára van szükségünk. Ezt a fehérjeszekvencia-tömeget már kezelni tudjuk egyrészt a hasonlósági modellezés, másrészt a "felcsavarodás-predikció" eszköztárával. Mind a két esetben léteznek olyan algoritmusok, amelyek a szerkezetpredikciót automatizáltan és rendkívüli áteresztõképességgel valósítják meg.
A Rockefeller Egyetem munkatársai egy cikkben leírták, hogy az élesztõ genomjából kiindulva az ott megtalált ismerõs, illetve ismeretlen fehérjéket képesek voltak "szerekezetprediktálni": néhány nap alatt több mint 1070 fehérje szerkezetét jósolták meg. Ezek a nagy teljesítményû predikciós szerverek komoly szerepet fognak játszani.
Az ígéretes kísérleti megközelítés a nagy áteresztõképességû röntgen-krisztallográfia.
Mit jelent a nagy áteresztõképesség? Hiszen nem a röntgenfelvétel idõigényes, hanem a kristályosítás.
Ma már létezik olyan automatizált berendezés, ahol a kombinatorikus kémiához hasonlóan a kristályosítást fel lehet gyorsítani. A kristályosítási körülményeket mátrixszerûen változtatják, így nagyon sok kristályosítási körülményt nagyon rövid idõ alatt képesek kipróbálni. Az amerikai National Institute of Health egyik projektjének az a célja, hogy négy éven belül 10 000 új kristályszerkezetet állítsanak elõ.
Ez a módszer egy kicsit hasonlít azokhoz a sakk-programokhoz, amelyek minden lehetõséget kiszámolnak, és ennek segítsévégel képesek megverni a világbajnokot.
Intellektuális kihívást természetesen az a feladat tartalmaz, amikor az ember végiggondolja, hogy a dolgok hogyan mûködnek, és ebbõl kiindulva igyekszik megoldást javasolni. A "nyers erõ" alkalmazása lényegesen kevesebb erõfeszítést kíván meg abban a stádiumban, amikor már mûködik. Amíg viszont létrejön a mûködõ állapot, rengeteg intellektuális kihívást tartogat. Ma már nem nagy teljesítmény, ha valaki egy nap kétszer elmegy Szegedre és visszajön. De ha naponta ezerszer szeretnénk megtenni ezt az utat, rögtön érezzük, hogy nem akármilyen feladatot kell megoldanunk. Ugyanez a helyzet a nagy áteresztõképességû tesztelésnél.
Általában felteszik, hogy ha két rendszer struktúrája hasonló, akkor a funkciójuk is hasonló. Ez azért érdekes, mert statikus párhuzamból következtetnek kinetikai analógiára. Milyen példák vagy ellenpéldák merülnek fel ebben az esetben?
A szerin-proteázok családja például elég nagy. Ezek a molekulák rendkívül hasonlítanak egymáshoz abban a tekintetben, hogy az aktív helyükön van egy katalitikus triád, amely a legtöbb szerin-proteázban azonos. Ezzel szemben a család tagjai különbözõ proteáz-funkciókat látnak el, aminek az a magyarázata, hogy sokszor az aktív helytõl távol esõ fehérjerészletek is szerepet játszanak a szubsztrát orientációjában, a pozicionálásában. Így a proteáz-aktivitás nem lesz teljesen azonos minden esetben.
Az általunk is sokat vizsgált citokróm P-450 enzimrendszer esetében inkább hasonlóságról beszélhetünk. Több mint 500 izoenzimet ismernek, és mindegyikben egy porfiringyûrû található az aktív helyen, amelyhez egy axiális cisztein kapcsolódik. Mindegyik izoenzim alapvetõen oxidatív átalakításokat végez a szervezetben, ennyiben a struktúra és a funkció összekötõdik, de ebben az esetben is tapasztalhatunk különbségeket, mert az a fehérje, amelyik befogadja ezt a porfirinrendszert, gyökeresen különbözik az egyes izoenzimekben, és ezért az izoenzimek nagyon különbözõ oxidatív reakciókat tudnak megvalósítani.
A bonyolult biológiai rendszereket gyakran nem "ceruzával megrajzolt" modelleknek próbálja megfeleltetni, hanem szerves kémiai rendszereknek. Milyen elõnyökkel jár ez a módszer?
Azért alkalmazunk biomimetikus modelleket, hogy azokat az oxidációs reakciókat modellezzük, amelyek a szervezetben például a citokróm P-450 enzimrendszer mûködése során megvalósulnak. Természetesen azt, hogy a szubsztrát hol oxidálódik, legalább két szempont határozza meg. Az egyik a szubsztrát elhelyezkedése az oxidációt végzõ centrumhoz képest. Ha ezt az orientációt nézzük, világos, hogy az átalakítást végrehajtó biokémiai rendszer mellett, amelynek a modelljét használtuk biomimetikus modellként, a fehérjekörnyezet is meghatározó jelentõségû. Ez rögzíti a megfelelõ pozícióban a szubsztrátot, és a szubsztrát ennek következtében csak egyféle módon illeszkedhet például egy zsebbe. Ebben a helyzetben találja meg az oxidációt végzõ rendszer, és az oxidáció bekövetkezik. A folyamatnak ezt a részét egyszerû preparatív modellel nem tudjuk modellezni, mert nincs meg az a fehérjekörnyezet, amely a természetes esetben a szervezetben ott van.
Ezeknek a folyamatoknak azonban van egy másik kritériuma is, mert hiába pozicionálja a fehérje a megfelelõ szerkezeti részletet az oxidációt végzõ porfirin irányába, ha az oxidáció nem következik be. Tehát van egy szerkezeti és egy elektronikus, a molekulák reaktivitásával összefüggõ szempont. Ennek a szempontnak a vizsgálatára viszont kiválóan alkalmas a biomimetikus modell: a modell alkalmazásával azt tudjuk megmondani, hogy egy szubsztrát mely pozíciókban oxidálódhat.
Ennek két nagyon fontos alkalmazási területe van. Egyrészt elõre jelezhetjük, hogy hol oxidálódik egy gyógyszerjelölt, tehát hol vannak az érzékeny pontjai. Ahhoz, hogy a gyógyszer a hatékonyságát a bevételt követõen sokáig fenntartsa, tehát ritkábban kelljen szedni, metabolikusan stabil jelöltre van szükség. Ezzel a vizsgálattal meg tudjuk mondani, hol metabolizálódik várhatóan a vegyület, és oda speciális, stabilitást növelõ funkciós csoportokat iktathatunk be. Tehát növelhetjük a stabilitását, ami elnyújtott hatást eredményez.
Másrészt a klinikai jelölt vizsgálatánál
felmerül, hogy ez az anyag egy élõ rendszerbe kerül.
Ekkor különbözõ mintavételi technikákkal
kimutatják, hogy mi történik vele, milyen metabolitjai
keletkeznek. Az engedélyezõ hatóság minden
egyes metabolit azonosítását megköveteli. Ehhez
sztenderdekre van szükség. Képzeljük el, hogy olyan
gyógyszerjelöltünk van, amely egzotikus helyen oxidálódik.
Hogyan tudjuk elõállítani a szükséges
metabolitot? Hagyományos oxidálószerekkel esetleg
nem tudunk ilyen oxidációt elõidézni, de ha
van egy biomimetikus rendszerünk, legalább az oxidáció
tekintetében alkalmas lehet a metabolit szintézisére.